Traditionally scientists have only been able to visualise and understand the genome in 2D but as we gather more data about how cells work it is clear that the 3D structure is extremely important for gene regulation and how cells differentiate. For example, a white blood cell looks and behaves differently to a red blood cell even though its genome is exactly the same.
Physically the genome is made up of several strands of DNA (chromosomes) that in total measure 2 meters long and have to fit into the cell nucleus which is approximately 1/10 width of a human hair. How does the cell machinery physically interact with such a complex and compact structure?
Why modelling the genome is important:
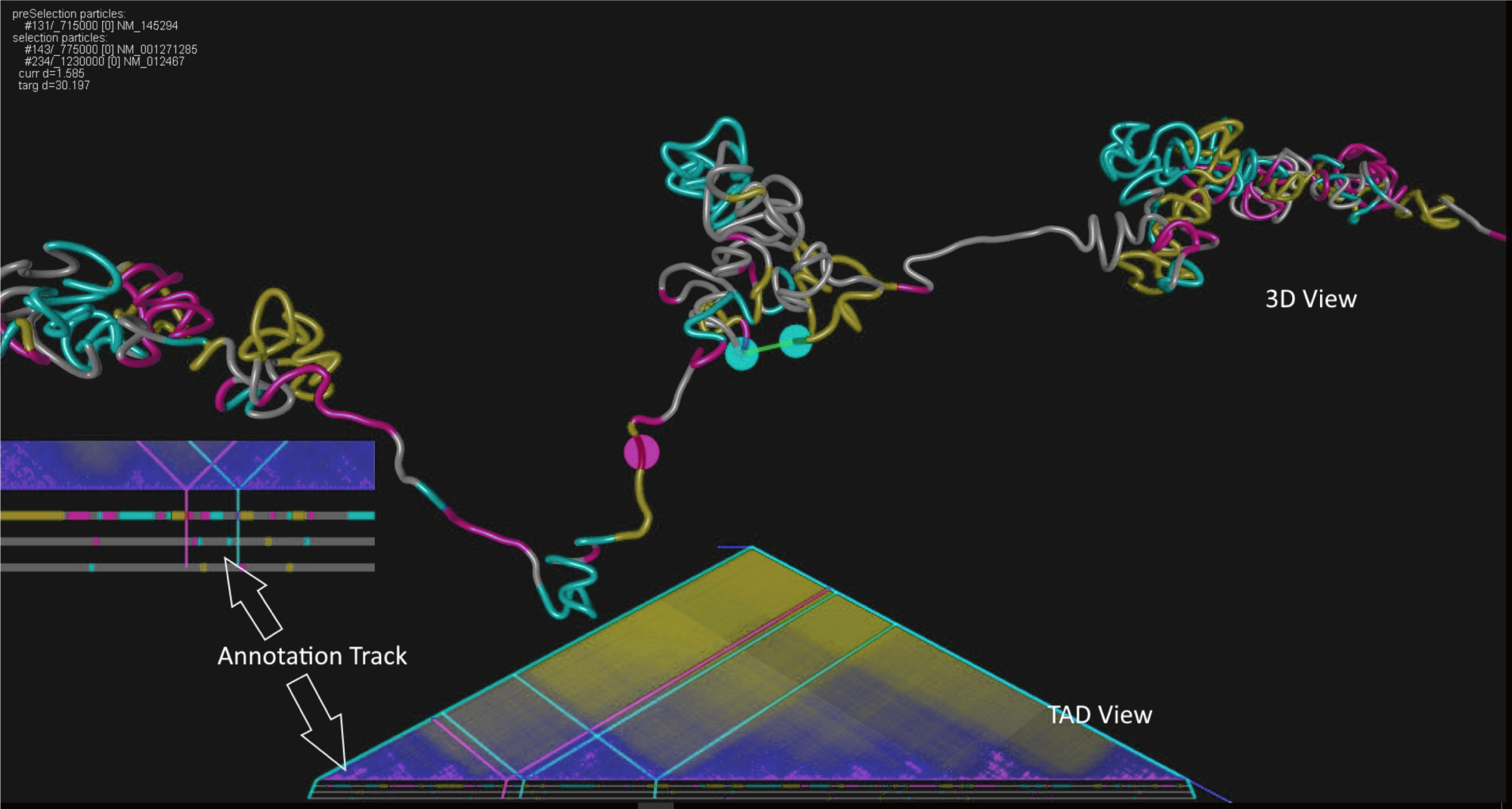
Recently scientists have begun to look at different groups of cells using so called ‘capture’ or ‘C’ techniques to look at where regions of the genome interact with each other. After some processing this produces a 2D interaction map which infers which part of the genome interacts with another part of the genome forming Topologically Active Domains or TADs.
However, these maps are not very intuitive to understand because we know the structure is in 3D. Steve Taylor (Computational Biology Research Group, MRC WIMM) and Frederic Fol Leymarie (Department of Computing, Goldsmiths University) began discussions after Steve saw Frederic presenting FoldSynth at BioVis and with Jim Hughes (Genome Biology, MRC WIMM), Stephen Todd (London Geometry) and Peter Todd (London Geometry) and William Latham (Department of Computing, Goldsmiths University) developed a prototype viewer called CSynth based on the 3D molecule viewer FoldSynth. They are seeking funding from various sources to continue develop this into a complete software package that also will allow import of public data, cutting edge 3D super resolution microscopy and detailed polymer models of the genome to get a much better understanding of how the genome is folded in cell and understand the complex mechanisms involved.
Even though the current prototype model of CSynth at this stage is not a ‘full’ modelling package (it uses simplified rules to model the DNA and currently does include any of the scaffolding proteins) it is allowing scientists to think about and pose questions more intuitively how and why genes, promoters and enhancers in different regions of the genome interact.